
近年来,多环芳烃(PAH)在多自由基领域的研究取得了诸多进展,但是设计合成具有非常规π磁性和多体强自旋纠缠的多自由基PAH 仍十分具有挑战。虽然通过在反应位点上添加取代基的方法,可以成功合成多自由基PAH,但取代基的存在会改变分子的化学稳定性和固有电子性质,不仅如此,溶解性差、反应性高都是其合成面临的困难。在开壳PAH中引入π磁性有如图1所示的两种原理,每一种原理都能够成功在开壳碳纳米结构中触发磁性:(1)通过子晶格不平衡,或一组蜂窝状二分晶格中的拓扑失稳(图1a);(2)利用π电子增强的电子-电子(e-e)相互作用,通过局域边界态的电子杂化与价电子之间的库仑排斥引入边界轨道的自旋对称性破缺(图1b)。然而,利用上述单一原理引入π磁性通常会限制相关自旋的数量或开壳层 PAH 中可达到的磁序类型,并且分子体系尺寸的增加也会给分子的稳定性和溶解度带来了重大挑战,阻碍了它们的制备。
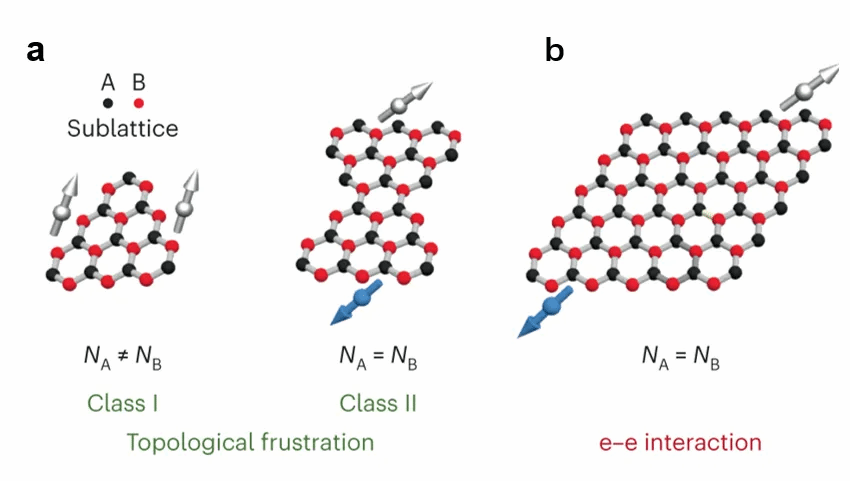
图1 单一π磁性起源概念原理。(a)拓扑失衡I类(也称为子晶格不平衡)和II类(两个子晶格均为拓扑失衡)导致的π磁性。黑点和红点分别代表子晶格A和B,而NA和NB表示子晶格A和B的数量。(b)e-e相互作用导致的π磁性。
在本文中,作者介绍了一种多自由基纳米石墨烯的设计方法,其具有多个强纠缠作用的量子自旋,这些自旋是由强e-e相互作用和拓扑失稳共同作用的结果(图2a)。作者通过将四个三角烯基序融合到菱形纳米石墨烯的边缘,构筑了一个蝴蝶形的纳米石墨烯,使得两个子晶格(A和B)在拓扑上均失稳,拓扑失稳使得蝴蝶形纳米石墨烯先产生了两个自由基(图2b)。此外,该设计还构筑了一个尺寸足够大、完全融合的开壳纳米石墨烯,通过强e-e相互作用触发占据前沿轨道的自旋对称性破缺,产生另外两个自由基。通过将上述两对自由基结合起来会得到一种四自由基纳米石墨烯,并且在该纳米石墨烯中同时存在铁磁和反铁磁耦合作用。为了进一步研究该蝴蝶型电子结构,作者通过电子紧束缚模型能谱对该分子电子结构进行了分析(图2c),该模型揭示了由于拓扑失稳分子存在两个零能量单占据分子轨道(φ2和φ3)和两个其他前沿轨道φ1和φ4,通过结合实验和理论计算,证实该系统包含四个不成对电子。
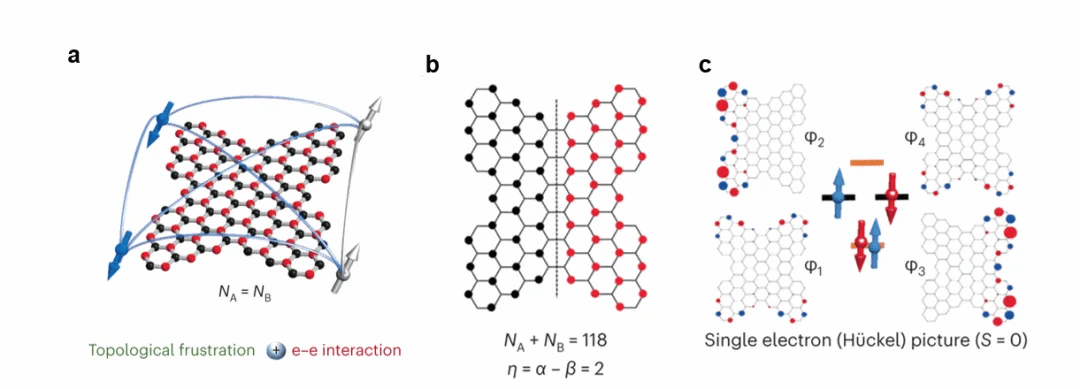
图2 强关联多自由基纳米石墨烯的概念设计原理。(a)具有不同磁性来源的开壳纳米石墨烯示意图。(b)分子1的“零点”说明,其中α和β分别指不相邻顶点和边的最大数量。(c)休克尔能谱显示两个零模式(φ2和φ3)和两个其他状态(φ1和φ4)。
蝴蝶形纳米石墨烯1是前体1′(图3a)在600 K 下Au(111)表面合成的,该前体由一个菱烯核心和四个通过锯齿状边缘连接到中心碳原子上的9-(2,6-二甲基苯基)蒽基结构组成,其键分辨STM(BRSTM)图像如图3b所示。作者接着进行了依赖于电压的微分电导光谱(IETS)测量,以探测局部电子结构,在分子1的角落和边缘处采集的dI /dV光谱均显示出两个分别位于-0.60 eV和0.75 eV能量处的特征峰(图3c),并且这些特征主要位于三角烯的角上和两个锯齿形边缘的海湾区域(图3e)。此外,相比于未占据态,占据态的特征在海湾区域中(在图3a中标记)表现出更高的dI /dV强度。为了更好地解释实验中的dI /dV图,作者利用完全活性空间自洽场(CASSCF)模型计算了戴森轨道,它描述了单个分子在电子电离/附着过程中电子分布的变化,从CASSCF戴森轨道模拟计算出的恒定高度dI /dV图(图3f)与实验恒定电流dI /dV图像(图3e)之间存在着良好的一致性。另外,角落处的IETS谱则在费米能级附近展现出对称的阶梯状特征(图3d),这归因于从基态到激发态的自旋翻转激发。CASSCF计算证实,除了基态(S = 0)之外,第一激发态为三重态,计算预测三重态(S = 1)的能量比基态高8 meV,与实验值 ~9 meV 几乎一致。为了进一步证实EF附近的IETS特征归因于自旋激发过程,计算了CASSCF 自然过渡轨道 (NTO)。NTO代表激发过程的紧凑轨道图像,在这种情况下,它们代表从单重态基态到第一个激发三重态的转变,图3h显示了模拟的自旋激发dI /dV图,该图对应于来自各个单线态和三线态NTO的贡献总和,与在9 meV下获得的实验dI /dV图(图3g)显示出良好的一致性,这进一步支持了在此能量下观察到的IETS特征源于自旋激发过程。
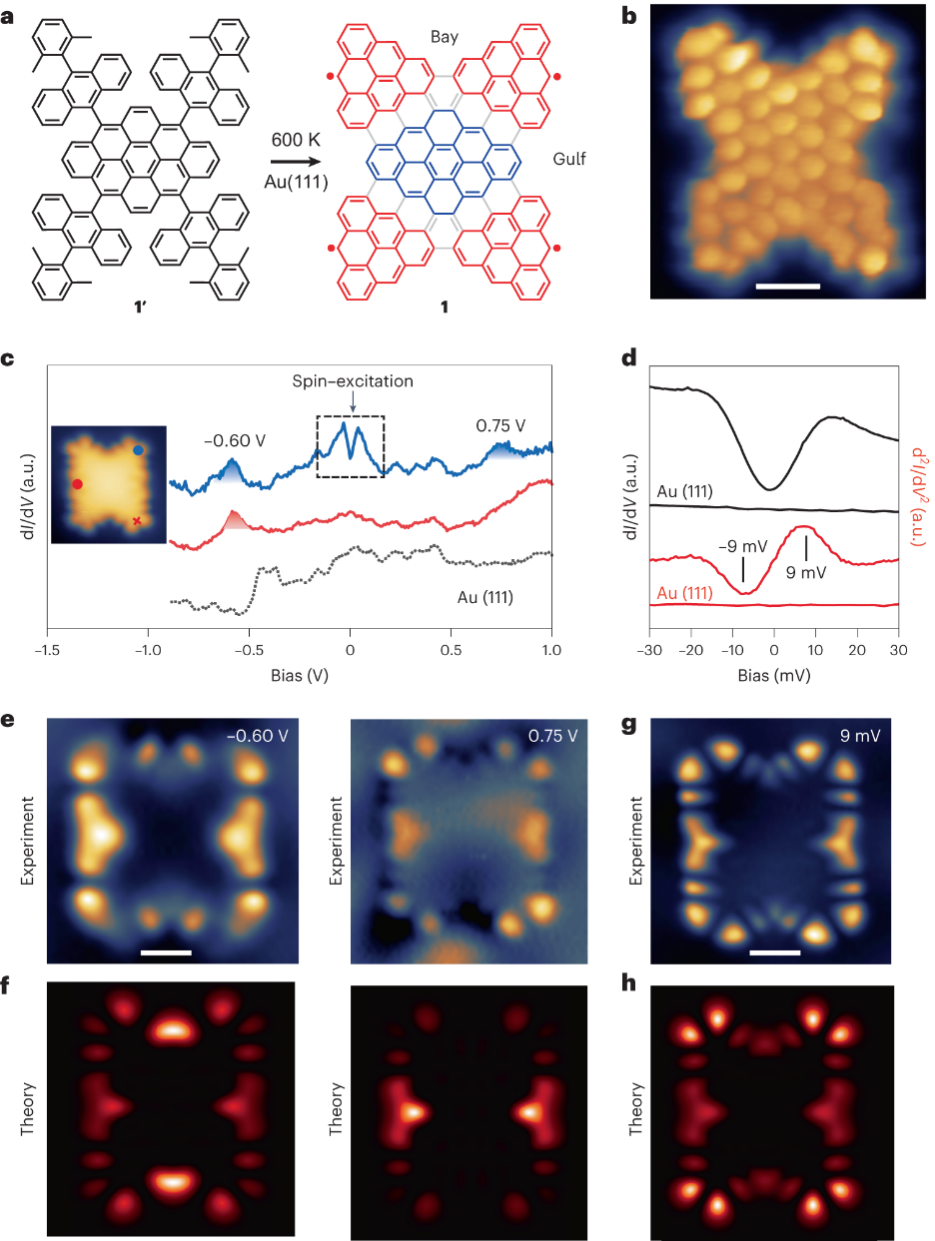
图3 分子1的表面合成与表征。(a)设计合成前体1′用于在Au(111)表面合成产品1。(b)用CO功能化尖端(Vs = 20 mV)拍摄的分子1的恒定高度BRSTM图像。(c)使用金属尖端在c插图中红点和蓝点标记的位置处获取1的点dI /dV光谱。(d)使用金属尖端在c插图中红十字标记的位置处获取1的点dI /dV和IETS光谱。(e)使用金属尖端在-0.6 V 和0.75 V 下拍摄的恒电流dI /dV图。(f)根据CASSCF戴森轨道计算的1的导带和价带上的恒定高度dI /dV图。(g)分子1的自旋激发图。(h)模拟分子1的自旋激发图。
为了确定分子1的基态特征并确认其四自由基性质,作者使用完全活性空间自洽场(CASSCF)方法进行了多体计算,并通过结合二阶N电子价态微扰理论在其基于域的局部,对自然轨道近似来解释CASSCF计算中缺失的电子相关性。从计算中可看出该纳米石墨烯的基态,(CAS(4,4))为拥有4个未配对电子的单线态,并由4个不同占据数的自然轨道组成(图4a),每个轨道的占据数接近1,这进一步验证了其四自由基特性。通过对其自旋-自旋相关性的理论分析可知(图4b),位于相同子晶格上的自旋表现出铁磁耦合,而位于不同子晶格上的自旋表现出反铁磁耦合。从图4b的插图可见,计算出的未配对密度主要集中在分子的四个角上,再次证实其四个未配对电子的存在。
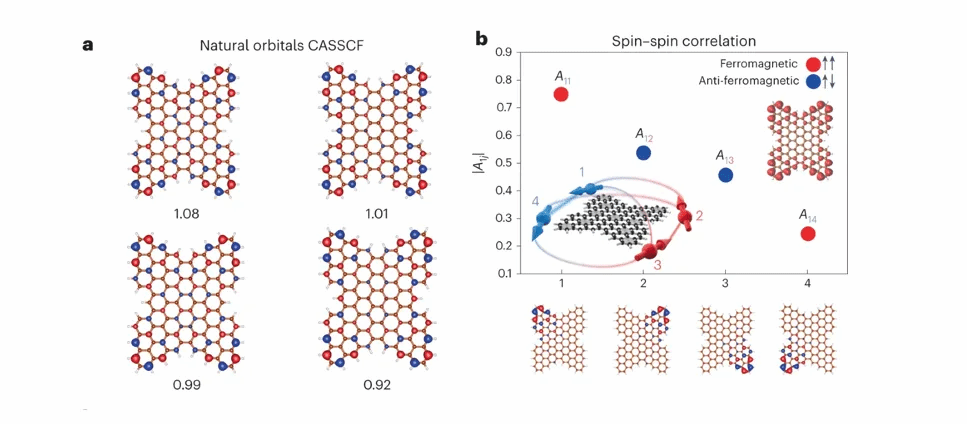
图4 具有开壳层四自由基特征的分子 1 的自旋相关分析。(a)CAS(4,4)中四个不同占据的自然轨道,每个自然轨道底部的数字代表占据电子。(b)通过理论分析计算出的自旋-自旋关联函数,左下插图为多自旋相互作用的概念图,右上插图为 CASSCF(4,4) 计算出的未配对密度分布,底部的四个轨道代表分子1各个角上自旋的局域化。
作者进一步采用了如图5a所示的扫描探针显微镜(SPM)技术和镍茂 (NiCp2) 功能化探针,通过获得不同尖端-样品距离的IETS光谱,直接验证了蝴蝶形纳米石墨烯的磁性能。如图5b在裸Au (111) 上拍摄的IETS光谱中,在对称于费米能级约±4 mV的位置存在相应峰/谷,表明NiCp2探针基态到双重简并激发态的跃迁。当探针接近Au表面时,峰或谷的特征能量保持不变。而在分子1的IETS光谱中(图5c),随着探针-样品距离减小,±4 mV处的信号向EF移动,而±13 mV处的信号远离EF,这种光谱的改变可以归因于NiCp2针尖与四自由基分子之间的磁交换耦合作用,这也为分子1中存在π磁性提供了直接的实验数据。
为了深入理解在分子1上获得的NiCp2探针的IETS光谱,作者设计了一个海森堡自旋模型。该模型由一个有效自旋哈密顿量组成,描述NiCp2探针和分子1自旋态之间的相互作用。分子的自旋模型由四个自旋位点组成,它们通过铁磁和反铁磁交换作用相互耦合,这由图4b中的自旋关联模型确定。此外,NiCp2探针和分子之间的相互作用由相关参数J ( z )控制,该参数随着尖端-样品距离 ( z ) 的减小而增加,进而通过精确对角化方法计算每个尖端-样品距离处模型哈密顿量的能谱。
图5d展示了当NiCp2尖端和分子之间的磁交换耦合强度(J)增加的情况下,模拟计算的自旋哈密顿量的IETS中不同自旋态的能级位置。当分子1和NiCp2都处于基态时,体系的能量最低,位于EF处。当J = 0 时,中心在± 5 meV的态(表示为A)对应于体系中分子1处于基态和NiCp2处于简并激发态,计算出该状态下的激发能量随着J的增加略有下降,这与5 meV处IETS峰的实验演变非常吻合,只有考虑NiCp2和分子1自旋位点之间的强烈不对称耦合才能出现这一特征。位于14 meV的态(表示为C)对应于体系中分子和尖端NiCp2都处于第一自旋激发态。值得一提的是,体系中分子的激发三重态和尖端的基态被预测位于±9 meV(表示为B),然而,这并没有在实验中观察到,可能是由于弹性隧穿电流极低,自旋激发信号无法检测到。
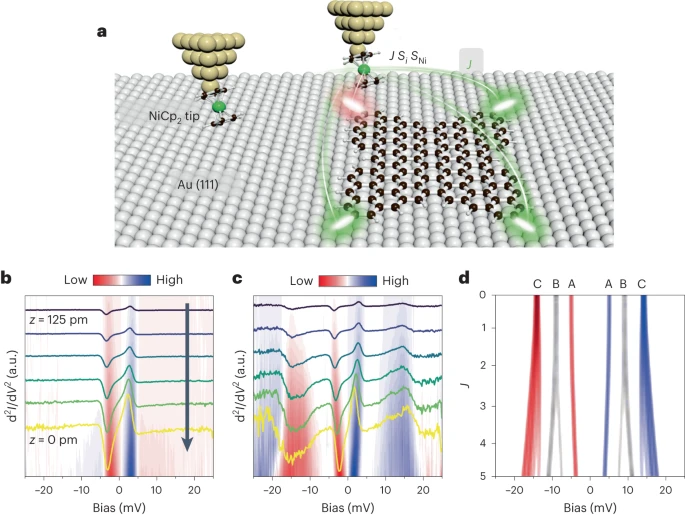
图5 使用NiCp2功能化尖端探测“蝴蝶”分子1的π磁性。(a)使用NiCp2尖端测量分子1的概念图。Si和SNi分别代表“蝴蝶”分子和 NiCp2尖端的自旋。J对应于分子和尖端之间的相互作用强度。(b)以色标绘制的在裸露的Au(111)上,并以25 pm的递减步长获得的IETS光谱。(c)以色标绘制在分子1的角落上,并以相同的25 pm递减步长获得的相应IETS光谱。(d)计算出的分子1的IETS光谱与耦合强度J的关系图。
文章通过融合强e-e相互作用和拓扑失稳的概念,设计了一种多自由基的蝴蝶形纳米石墨烯。该蝴蝶形的四自由基分子表现出高度自旋纠缠的多体基态。文章提出的这种多自由基强关联自旋的设计策略,为创建具有多个纠缠自旋的稳定量子单元开辟了一条新途径,作为构建可扩展和复杂量子网络的基本构件,这对于未来量子信息技术的发展至关重要。

编辑:刘玉研
")
