拉曼与热电小组
Nat. Mach. Intell. 2020,10,1038
Nature Machine intelligence: 高斯嵌入应用于大规模基因集分析
Gaussian embedding for large-scale gene set analysis
Sheng Wang, Emily R. Flynn and Russ B. Altman
前言
生物研究中通常会产生高通量的数据集(high-throughput data),包括大量的蛋白质结构数据、基因集数据等。其中,基因集表征着受其调控的蛋白质复合物的复杂信号通路(pathway),是生物研究的关键一环。因此,引入数据分析方法对大量基因集序列进行识别和分类,并分析其产生作用的信号通路有助于深入了解生物调控机理,是重要的研究内容。
网络嵌入(network embedding), 又叫做网络表示学习, 用于将研究对象表示为结构化的网络,并分析对象间的网络化联系。机器学习能够有效的处理大量的向量数据, 而网络数据作为最普适、广泛的数据形式之一, 对其进行有效表示,并运用成熟的机器学习算法来分析网络数据也成为近几年数据挖掘和机器学习的研究热点。
近日,斯坦福大学 Russ B. Altman课题组提出了一种基于网络的高斯嵌入算法Set2Gaussian,用于分析高通量基因集数据。该方法将大量基因嵌入到低维度空间中,并将基因集示为低维度空间的多元高斯分布,由此分析基因间的相互联系。算法得到的基因集嵌入表示(gene set embedding representation)被用于识别基因集中的成员基因,划分癌症的亚型,寻找简明基因集等三个场景,均取得了比传统方法显著的识别效果。
内容
文章首先介绍了高斯嵌入算法Set2Gaussian,而后在三个不同场景下测试了这种新的嵌入算法进行基因组识别和分类的表现。Set2Gaussian算法描述如下:
Set2Gaussian高斯嵌入以生物网络及一系列的基因集作为输入量,输出每个基因集的多元高斯分布。
首先,算法设置了嵌入网络的结构。Set2Gaussian定义了n*n维(n表示全部基因的个数)的邻接矩阵A,用于存储基因集中各基因的连接关系,以此表示基因间信号通路。同时定义向量V用于存放顶点(即基因集中的各个基因),相当于用图表示基因集 ,顶点为单个基因,边为信号通路,矩阵H表示所有的基因集,并设置了m种基因集,即有m个子网络图。信号通路(pathway)描述了基因对蛋白质作用的机理,如原材料被a基因编码的蛋白质1转化为产物A,再被b基因编码的蛋白质2转化为产物B,将涉及信号通路的基因按先后顺序连接起来,组成构建嵌入网络的有权向量图。子网络结构进一步借助随机游走(random walk)方法优化(图1左)。
其次,算法对基因集进行数学化表示,并嵌入网络。每个基因被嵌入在一个低维空间中,转化为低维度的数据点,同时每个基因集表示为多元高斯分布,据此识别不同的信号通路(图1右)。多元高斯分布借助最小化损失函数实现,损失函数依据以下两个准则:(1)具有类似扩散状态的基因应该彼此接近,(2)基因在隶属的基因集中应该有更高的高斯分布概率。
总体而言,该算法设计并优化嵌入网络,由此输出基因的低维度向量以及基因集的多元高斯分布,借助输出结果识别隶属于不同基因集的基因及其信息通路,进而为各生物要素的聚类和分型提供依据。
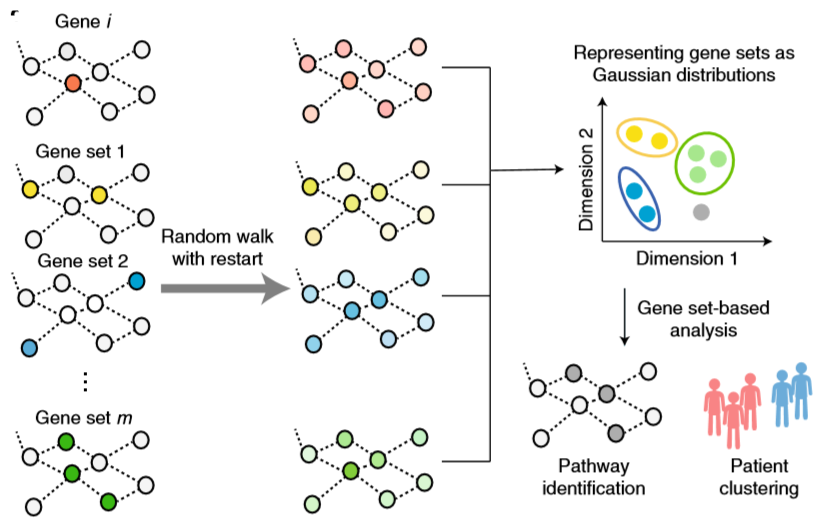
图1. 网络嵌入算法示意图
接下来,文章介绍了Set2Gaussian算法的三个应用场景,并与其他传统方法对照:
在先前研究中,同一基因集中的基因被一起分析,以研究潜在的生物学过程。然而,高通量实验产生的基因集可能包含大量的假阳性和假阴性基因。因此,借助识别基因组成的员基因来降低噪声是大规模基因分析的重要任务。
文中,包含Set2Gaussian算法在内的四种方法被用于识别三个基因组数据集中的基因组成员,识别结果借助精确率-召回率曲线(AUPRC)评估,该方法运用识别准确率评估方法优劣。图2所示结果表明,Set2Gaussian嵌入网络在三种数据集中均比基线方法更好地将基因归入基因组。如图2b所示,在Reactome数据集中,对于11-30个基因,Set2Gaussian算法在AUPRC以下的面积为0.48,优于mean(0.42)、weighted mean(0.40)和max(0.22)方法。

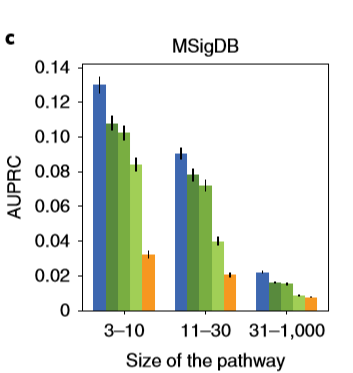
图2. Set2Gaussian识别基因组成员及结果评估 a-c, 在NCI(a)、Reactome(b)和MSigDB (c)数据集中,Set2Gaussian与其他四种方法识别基因集成员的比较。
肿瘤分类将不同种类的肿瘤分为临床和生物学上有意义的亚型,突变基因集是肿瘤分类的重要指标。文章选取肿瘤突变基因数据集,运用Set2Gaussian把基因集表示为多元高斯分布并进行聚类,同类肿瘤被归入相似的基因组通路中,分类结果与其他三种方法进行比较。借助Kaplan-Meier生存分析方法(KM)评估聚类结果,该方法运用存活率评估数据集中是否存在显著差异。如图3a所示,在Set2Gaussian方法中,新的亚型在各组间的存活率指标有显著差异,而三种对照方法则没有显著差异。图3b,d详细列举了Set2Gaussian方法的KM检验结果,两种亚型均在生存率上表现出显著的差异。
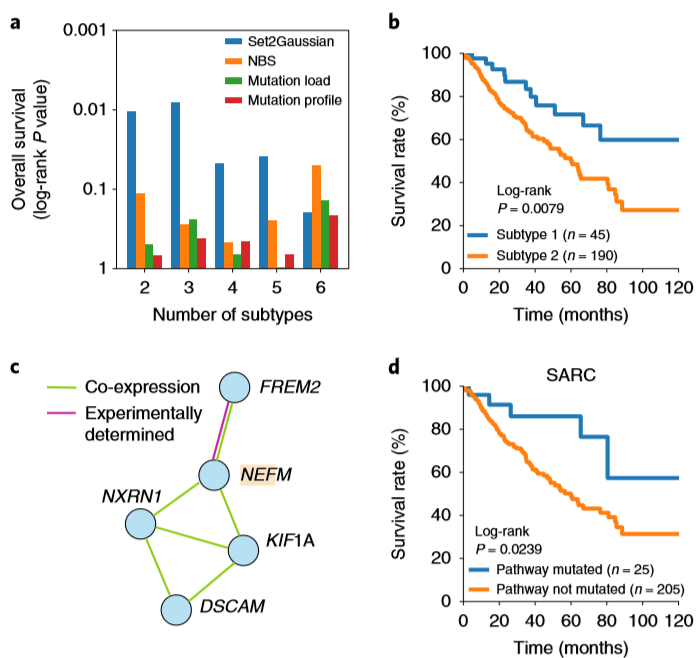
图3. Set2Gaussian识别新亚型的聚类及KM检验结果 a, 在235种不同亚型的肉瘤肿瘤分层中,Set2Gaussian和其他方法的比较。b, KM图评估Set2Gaussian方法,使用所有基因将肿瘤聚类为两种亚型。c, 在肉瘤中Set2Gaussian识别的子网络。d, KM图评估Set2Gaussian方法,仅使用子网络聚类。
在生物基因研究中,GSEA被广泛用于从MSigDB等基因集数据库中寻找显著富集的基因集。虽然GSEA提供了可能富集的基因集列表,但随着基因集数据库的不断增长,该方法不可避免地会导致预先定义的基因集中存在大量冗余信息,冗余降低了富集分析的速度。因此,减少预先定义的基因集数量,只保留最具信息性和代表性的简明基因集,将具有巨大的潜在价值。
文中Set2Gaussian方法被用于寻找简洁集,并与其他四种方法进行比较。如图4a所示,分析结果表明由Set2Gaussian方法发现的简明集效果最好。总体来说,Set2Gaussian方法中72.1%的查询基因组被显著性富集,高于All方法的68.5%,标准方法的50.5%,比例方法的16.4%,及hiting方法的15.1%。文章继续列举了详细的Set2Gaussian方法与传统方法的性能对比。图4b,c进一步对比了Set2Gaussian方法和两种对照方法在显著性富基因集占比上的差异,图4d分析了显著富集基因在Set2Gaussian和All对照方法上的KM检验的结果,这些评估手段均进一步证实了Set2Gaussian方法在准确率和显著性上优于传统方法。
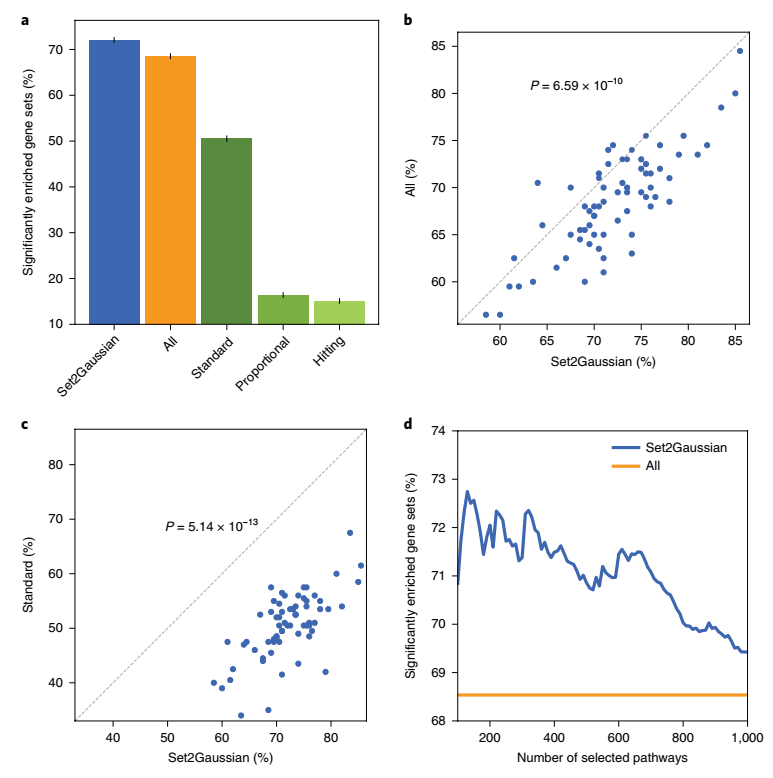
图4. Set2Gaussian法在寻找简明基因集中的应用及KM检验结果 a, 在GSEA中使用五个不同的先前定义的基因集的比较。b, 使用之前定义的set2Gaussian过滤的基因集与使用所有之前定义的基因集的比较。c, 使用预先定义的set2Gaussian过滤的基因集和使用预先定义的标准覆盖集衍生的基因集的比较。d, KM检验,由Set2Gaussian选择的预先定义的不同数量的基因集组成的显著丰富的基因组的数目。
总结
嵌入网络是研究结构化网络的算法,它将神经网络与有权向量图结合起来,使之能分析大通量数据的相互关系。本文设计了基于网络的高斯嵌入,将基因集表示为多元高斯分布,利用这些多元高斯分布得到了更高质量且紧致的数据特征,作为机器学习的输入,优化基因集的识别。本文所涉及的方法也可以推广至具有大量相关性特征的研究领域,如药物合成,反应机理等。
原文链接:https://www.nature.com/articles/s42256-020-0193-2
源代码及数据链接:
https://doi. org/10.5281/zenodo.3827929
https://doi.org/10.6084/m9.figshare.11341181.v1
撰稿人:唐永翔 (2018级硕士生)
校稿人:林禄春 (2018级博士后)
")
