电化学与单团簇小组
Nano Letter:利用末端乙炔与银电极的相互作用形成Ag-C键构筑分子结
Covalent Ag-C Bonding Contacts from Unprotected Terminal Acetylenes for Molecular Junctions
Songsong Li, Hao Yu, Xinyi Chen, Andrew A. Gewirth, Jeffrey S. Moore, and Charles M. Schroeder*
前言
分子-金属接触界面的性质对于控制分子结的电输运性质一直扮演着重要的作用。实现具有高鲁棒性和低接触电阻的有机分子-金属电极连接对于构筑高性能的单分子电子器件至关重要。当前分子与金属电极的连接主要是基于修饰在分子上的锚定基团与金属电极的相互作用,在这些锚定基团中通过共价键锚定(巯基)比通过配位键锚定(氨基、吡啶、恶唑)的接触电阻通常要小10到100倍,所以巯基锚定是常用的锚定基团。但是基于巯基锚定也会有一些弊端,比如巯基在金属界面表现出的瞬态物理吸附/解吸,从而对单分子器件的稳定性产生影响。
在本篇文章报道之前,Olavarria-Contreras(J. Am. Chem. Soc. 2016, 138, 8465.)以及Bejarano(J. Am. Chem. Soc. 2018, 140, 1691.)等人分别报道了在不需要化学保护基团的情况下,使用乙炔锚定形成金-碳(Au-C)共价键来构筑分子结的方法。但是该方法有两点问题:一是对于小分子来说利用该方法形成稳定分子结的效率较低;二是基于末端乙炔形成Au-C键的分子结电导比基于sp3碳形成Au-C键的接触电阻要大。
针对以上问题,作者设计了一种基于末端乙炔与银电极相互作用形成Ag-C键来构筑分子结的方法,通过该方法构筑的分子结具有高鲁棒性、高导电性以及不需要巯基锚定等的特点,拉曼光谱以及单分子电导测试证实了Ag-C共价键的形成,DFT理论计算进一步揭示了分子与金电极的连接构型。
内容
在该研究中,作者首先利用扫描隧道显微镜裂结技术(STM-BJ),使用银电极对目标分子P1-P3的电导进行测试(Fig1 a),得到了目标分子的一维电导统计图(Fig1 b)和二维电导强度图(Fig2 a-c)。为了进一步理解金属电极在电输运中的作用,作者还采用常规的金电极对三个分子的电导进行测量(Fig2 d-f)来作为对比,发现通过Au-C键构筑的分子结比Ag-C键构筑的分子结有更强的电输运能力。
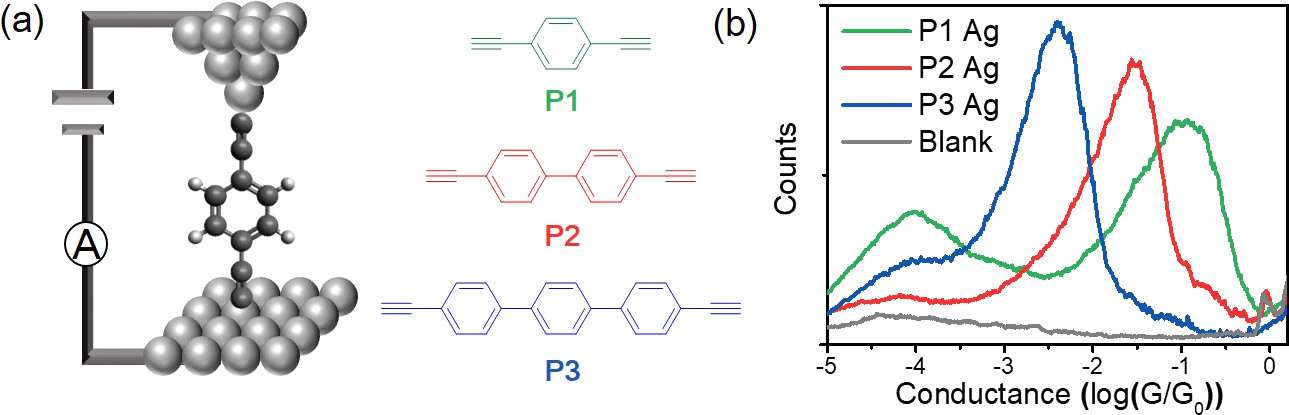
Fig1 a,电输运测量示意图以及三种乙炔末端分子导线P1-P3的分子结构;b,利用银电极测量目标分子得到的一维电导统计图。
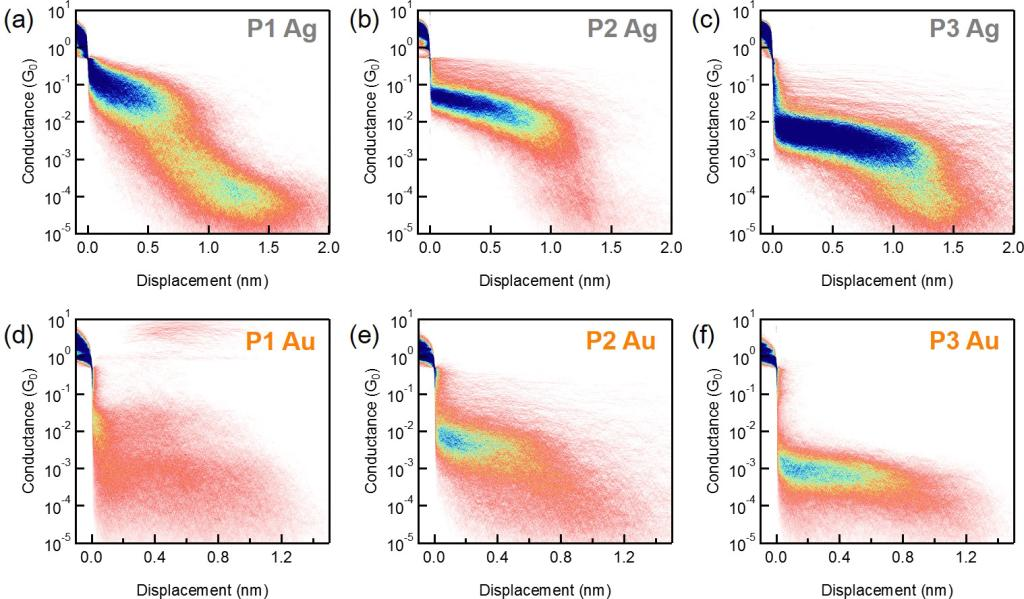
Fig2 a-c,利用银电极测量P1-P3分子得到二维电导强度图;d-f,利用金电极测量P1-P3分子得到二维电导强度图。
作者把三个分子分别采用-CH2-Au,-C≡C-Ag以及-C≡C-Au三种连接测得的分子电导对应分子长度做图。由量子隧穿模型可知G/G0 = A exp (-βL),其中A为接触电阻,β为分子衰减常数,L为分子股价长度。通过对同种末端连接的系列分子P1-P3的电导-长度连线,并反推至L=0处即可得到该分子与界面的接触电阻A。经过计算发现,-C≡C-Ag分子结的接触电阻为6 KΩ,-C≡C-Au分子结的接触电阻为36 KΩ,对比巯基锚定的40 KΩ和氨基锚定的189 KΩ, 由此可得出以Ag-C键构筑的分子结接触电阻明显小于Au-C键连接以及巯基锚定的接触电阻。
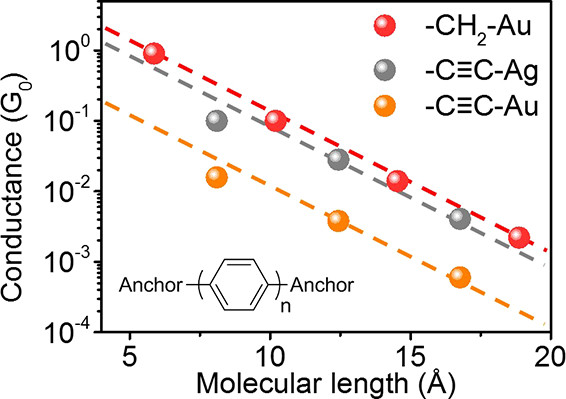
Fig3 三个目标分子P1-P3分别采用-CH2-Au,-C≡C-Ag以及-C≡C-Au连接得到的电导-长度散点图。
如图Fig4所示,作者通过将P1-P3分子分别组装到金基底和银基底上来测表面增强拉曼光谱,其中1952 cm-1和2117 cm-1处的峰分别归属为苯环和碳碳三键。先前的一些工作已经证实,如果碳碳三键上的碳以共价键锚定在金属界面上,则2117 cm-1处的峰会转移到2000 cm-1处,并且峰会变宽(J. Am. Chem. Soc. 2012, 134, 19425.)。所以在上图中也可以看到2000 cm-1处的峰可以分别指认为来自Au-C键和Ag-C键。
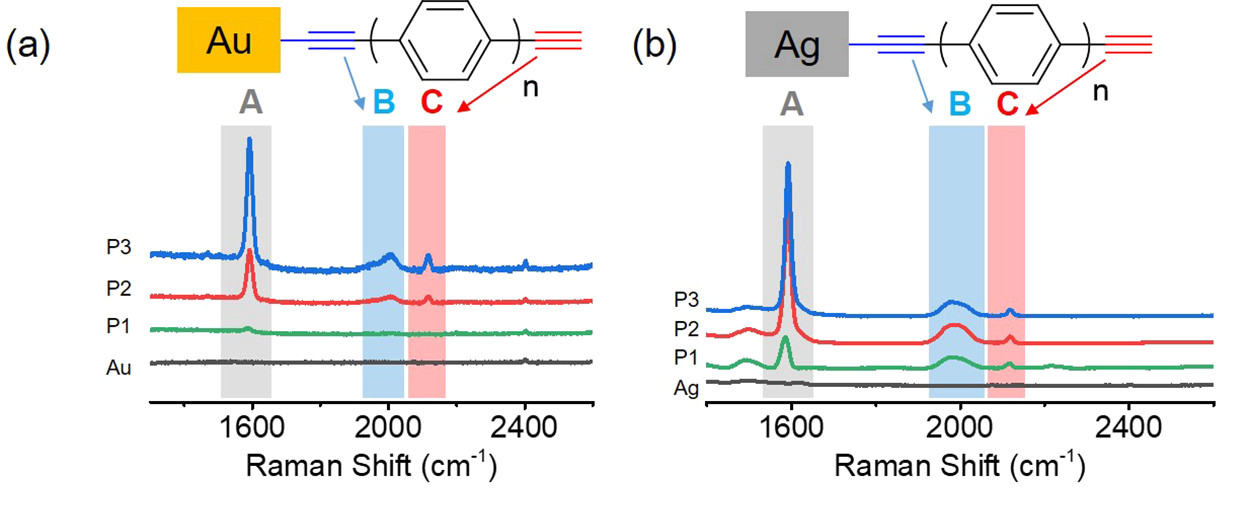
Fig4 P1-P3 分子分别在金基底(a)和银基底(b)上测得的表面增强拉曼光谱(SERS)信号。
为了进一步理解采用金电极和银电极测量时P1-P3分子电导的差异,作者采用非平衡格林函数-密度泛函理论(NEGF-DFT)进行了理论模拟。如Fig5 a所示,模拟 发现基于Ag-C键的分子结在费米能级处采用atop的连接构型的分子电导小于基于Au-C键的分子结的电导,但是基于Ag-C键的分子结在费米能级处采用Hollow的连接构型的分子电导则大于基于Au-C键的分子结的电导,后者与实验结果一致。Fig5 b分别展示了在Hollow和atop构型中分子在金电极上的连接位点。
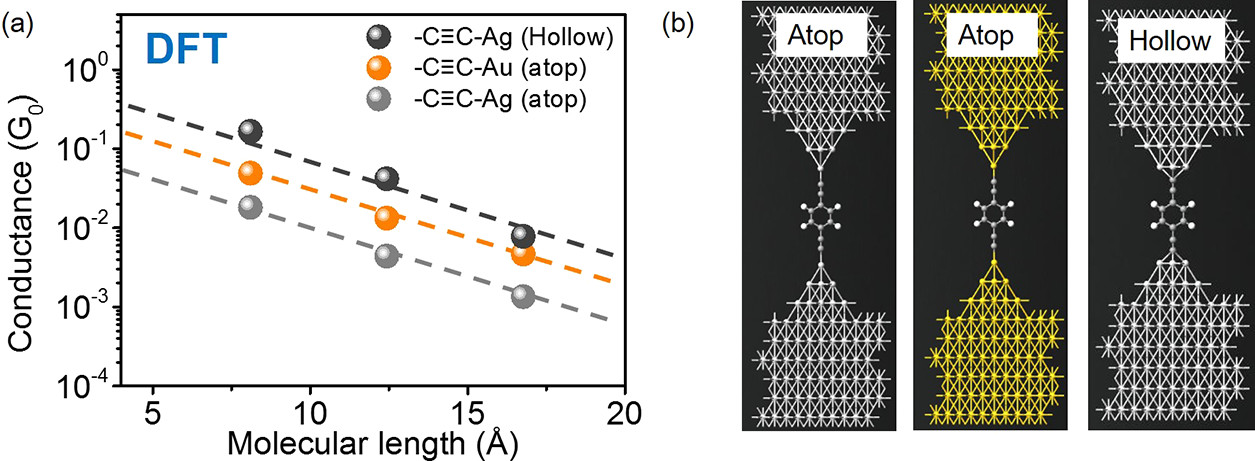
Fig5 a,对目标分子分别在Hollow, atop连接构型采用密度泛函理论(DFT)进行模拟;b,采用NEGF-DFT对P1分子的连接构型进行模拟。
通过之前的工作我们知道,当两个连接位点在苯环上的间位时,就会存在相消量子干涉效应(DQI)使得分子的电导减小;相反如果连接位点在苯环上的对位时,就会存在相增量子干涉效应(CQI)使得分子的电导增大(Nature Reviews Physics 2019, 1, 381.)。通过实验,作者发现通过银电极测量的对/间位P1分子的电导比为Gpara/Gmeta = 69,通过比较氨基(Gpara/Gmeta =2)/巯基(Gpara/Gmeta =10)和硫甲基(Gpara/Gmeta =2)的电导比,可以发现银电极在调控量子干涉方面的巨大潜力,这为设计具有更大电导比的单分子晶体管提供了新的思路。
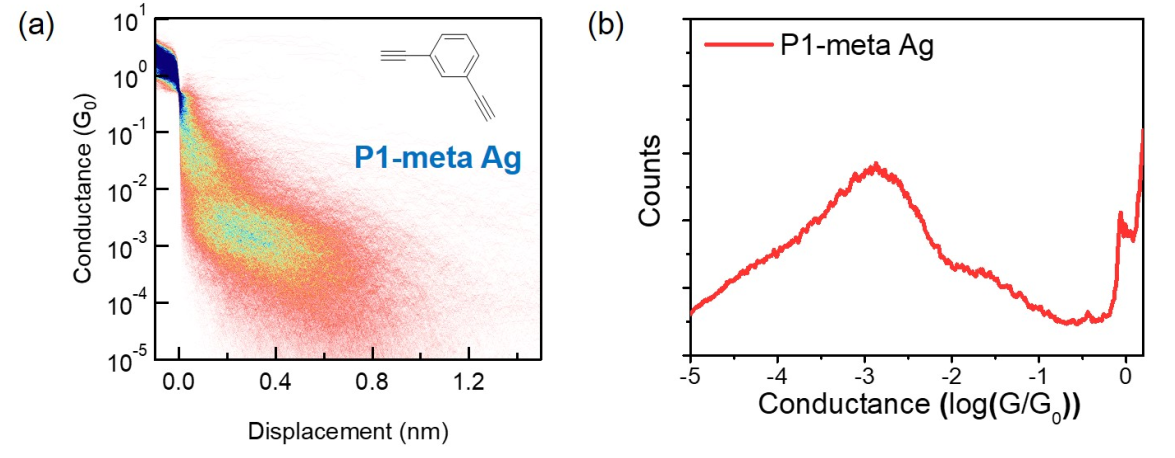
Fig6 a-b, 间位P1分子的二维电导强度图以及一维电导统计图。
原文链接:https://doi.org/10.1021/acs.nanolett.0c02015
撰稿人:李瑞豪(2018级博士生)
校稿人:袁赛赛(2018级博士后)
")
