电化学小组文献分享
Chem:同性电荷不一定相互排斥?碘鎓离子和银离子两个正离子之间的相互作用

卤素原子失去一个电子形成卤素阳离子X+ (X = I, Br, Cl)。在卤键(XB)中,X+被认为是强的卤键受体,它可与两个卤键给体(如吡啶)形成一个[N-I-N]三中心四电子的双(吡啶)碘鎓离子配合物。本研究发现通过卤键形成的配合物[L1-I-L1]+和[L2-Ag-L2]+配合物之间,存在I+和Ag+相互作用,即带正电荷的两个阳离子克服静电排斥发生了相互作用,其中碘阳离子为亲核试剂向银(I)阳离子提供电子。X射线衍射分析表明,I+和Ag+之间距离较短[3.4608(3) Å]。另外,等温滴定量热法检测结果表明,两个配合物比为1:1时,结合常数为37,000 M-1(ΔH = 5.170 kcal/mol,ΔS = 3.56 cal/mol,ΔG = 6.231 kcal/ mol)。作者进一步采用DFT(密度泛函理论)对I*Ag相互作用性质进行研究,结果表明,I+具有亲核性,而且I+和Ag+阳离子之间表现出异常的强吸引作用。
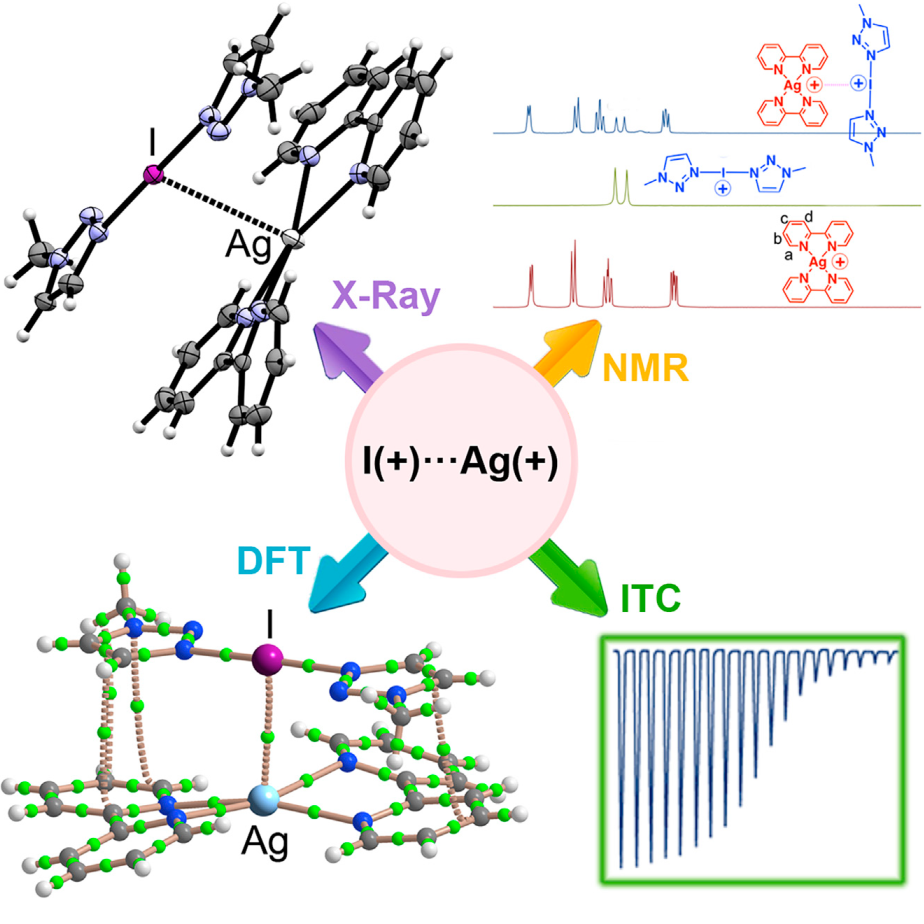
TOC:I+和Ag+阳离子的相互作用
研究背景
自2007年以来,卤键的研究经历了爆炸式增长,已成为近20年来研究最多的非共价相互作用之一。卤键被定义为发生在卤素原子表面静电势为正的区域(具有亲电性)和中性或阴离子的路易斯碱之间的相互作用。因为其存在本身具有争议性,所以在采用众多非共价相互作用指导超分子组装的研究中,卤键在很长的一段时间内被忽略。但卤键与普遍存在的氢键的区别是非常明显的。尽管如此,卤键现已被用于控制多种主客体系统的自组装,其应用范围从多孔材料、液晶、磁性、磷光材料到离子对识别、生物分子工程和化学分离。卤键因其方向性、特异性和高强度在当代晶体工程中蓬勃发展,基于卤键制备的高度复杂的结构具有很高的精度和准确度。尽管卤键相对于氢键具有更强的方向性,但它对环境更敏感,因此大部分卤键因其存在于固体中而被证明。
当卤素极化到电子完全从卤素原子移走的极限时,就会产生带正电荷的离子,即卤素阳离子X+ (X = I, Br,或Cl)。因此,卤素离子(阳离子)可以被认为是极端极化的卤素原子,因此属于卤键的领域,是非常强的卤键给体。然而,由于这类卤素离子的反应活性较高,使其与其他经典的卤键给体显著不同。Hassel等人在20世纪60年代第一次报道了存在于双碘(吡啶)配合物中的卤素阳离子,并证明其存在[N-I-N]+三中心四电子结构 [1] 。在首次报道标志性的卤素阳离子配合物近30年后,20世纪90年代Barlueng等人发现了一种温和的碘化试剂和氧化剂,双(吡啶)碘鎓(I)四氟硼酸盐,也就是今天的Barluenga试剂[2]。Barluenga试剂是一种相对稳定的白色固体,可溶于有机和水溶液,其在有机合成中的适用性已得到广泛论证[3]。 [N-I-N]+卤素键在溶液中的对称性已被研究证明[4-9],最近以卤素为基础的二氧碘烷配合物也被用作有机合成的试剂[10]。
基于卤素阳离子的卤键配合物是通过[N…Ag+…N] →[N…I+…N]进行阳离子交换而制备,卤素阳离子通常结合两个相同的XB受体,形成线性和均配卤素配合物,本文报道了第一个杂配的(不对称的)卤素配合物的合成,在固态及溶液中的特性,该配合物表现出非常不寻常的和显著的阳离子之间(从I+到Ag+)的吸引作用,与众所周知的亲银相互作用不同,碘鎓离子和Ag+之间的相互作用至今并未被报导,计算研究表明,碘离子…银(I) (I…Ag+)相互作用与类似的亲银离子(Ag+…Ag+)相互作用无关,而与I+的亲核特性有关。
合成及表征
一价银配合物2,4,6及碘鎓配合物5和7的合成路径如图1所示,配合物7 [I(mtz)2]PF6*[Ag(bpy)2]PF6 是由配合物2和5以1:1比例在DCM(二氯甲烷)中混合缓慢挥发溶液而得到,分子中I+…Ag+发生相互作用(用I*Ag表示)。所示配合物7可通过三种方法得到(如图1),所有中间配合物可通过单晶X衍射进行表征。
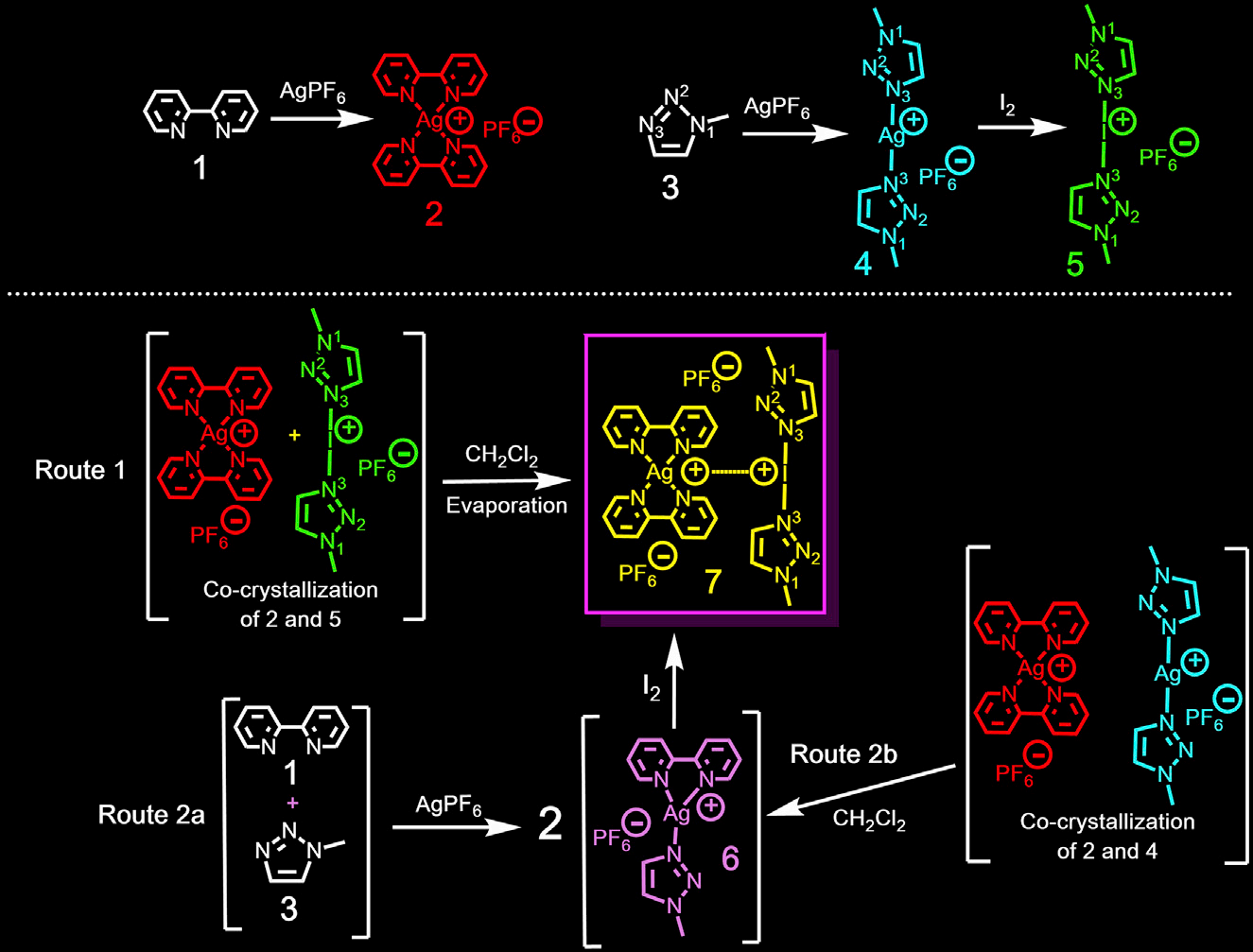
图1:配合物2、4、5、6的合成,以及配合物7的三种不同的路线
7的三种不同的制备方法都得到了相同的1H NMR谱(图2A),对比2、5、7的1H NMR谱图(图2A)可以看出,配合物7的三唑质子与配合物5的相应信号相比没有变化。然而,由于配合物7在溶液中持久存在的I*Ag相互作用,7中bpy(2,2,—联吡啶)的质子(a, b, c, d) 与配合物2相比表现出可检测的化学位移变化,这可能是由于I*Ag相互作用减弱了配合物2中的Ag-N相互作用,增加了氮原子周围的电子密度,从而导致联吡啶环Π电子密度的重新分布。接着作者还紫外可见光谱(图2B)和荧光光谱(图2C)对其进行了证明,结果与核磁结果一致。为了研究I*Ag相互作用的强度,作者利用等温滴定量热法在二氯甲烷中表征了它的结合常数为37,000 M-1(ΔH = 5.170 kcal/mol,ΔS = 3.56 cal/mol,ΔG = 6.231 kcal/ mol),这些结果证明在溶液中存在较强的I*Ag相互作用。
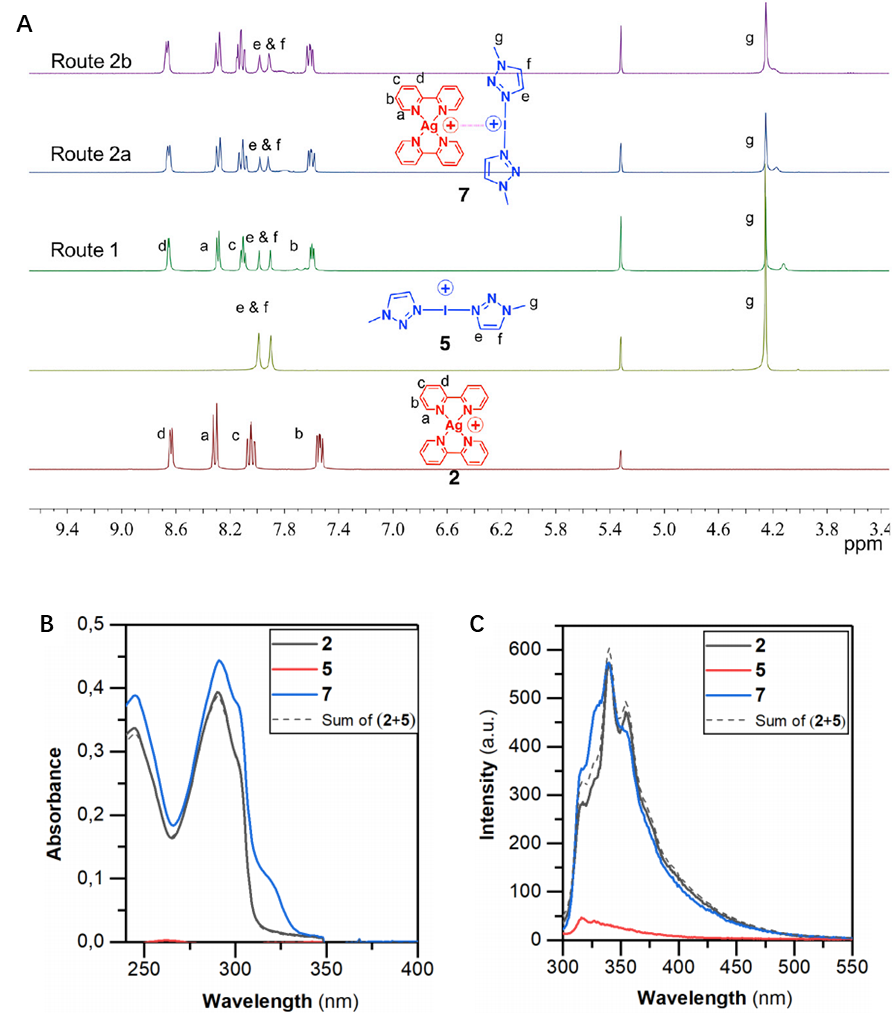
图2:配合物2,5及不同合成路线得到的配合物7在CD2Cl2中的1H NMR谱图(A);配合物2,5,7及2,5混合物的紫外光谱(B)及荧光光谱(C,λex=290 nm),虚线为2和5的理论组合谱。
在固态中,I*Ag相互作用是由配合物7中的 [Ag(bpy)2]PF6 (2) 和[I(mtz)2]PF6 (5)两部分组成,在[I(mtz)2]PF6的两个三唑环和[Ag(bpy)2]PF6的两个吡啶环之间形成交替偏离中心的(…I*Ag…I*Ag…I*Ag….)Π堆积共面阵列(三唑环-吡啶质心到质心的距离= 3.540,3.675,3.738和3.771 Å)。相邻的[Ag(bpy)2]PF6中两个吡啶环进行对应横向交联 (吡啶-吡啶质心到质心的距离=4.898和5.031˚)。由单晶X射线衍射表征结果可得Ag+和I+之间距离是3.4608(3) Å,该距离小于两元素的范德华半径之和(3.70 Å),也比配合物2中Ag+…Ag+(3.5406(8) Å)的距离短(图3)。
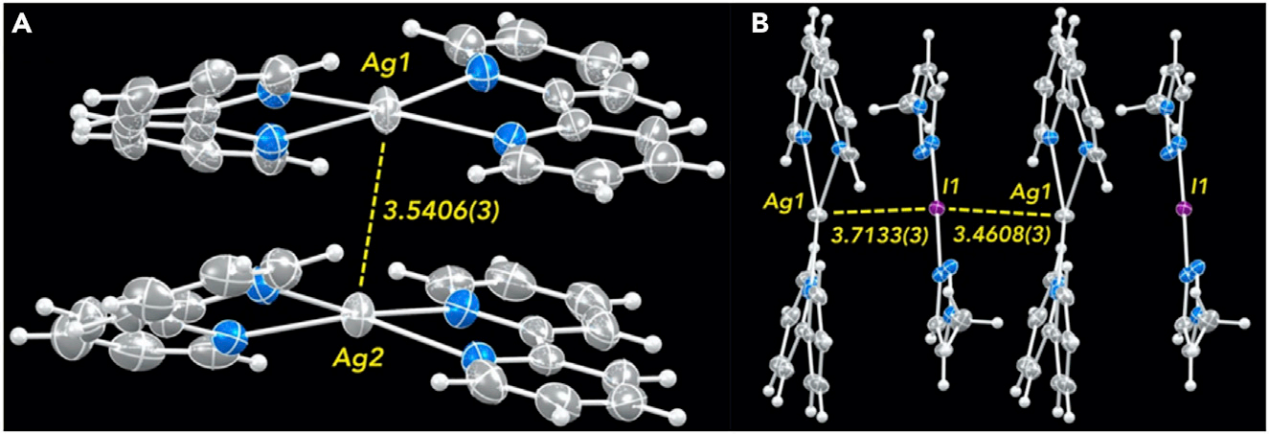
图3:配合物2 (A)和配合物7 (B)的x射线晶体结构
理论计算
DFT计算的目的是比较在化合物2的X射线晶体结构中观察到的亲银Ag+…Ag+相互作用和化合物7中的I+…Ag+相互作用,特别是理解7中从未观测到的的I*Ag非共价相互作用的物理性质。首先,对2的二聚体和7的二聚体(Ag+…I+距离最短的二聚体)进行了分子中原子的量子理论(QTAIM)分析。图4A,B给出了两种化合物的键临界点(BCPs)分布和键合路径。对于2的二聚体(图3A),分布显示了BCP的存在(表示为绿色球体)和连接Ag原子的键路,确认了亲银相互作用的存在。此外,几个BCP和键合路径将配体的几个C/N原子相互连接,从而证明了p-p堆叠作用的存在。同样,在化合物7中,I*Ag相互作用的特征是一个BCP和连接Ag原子和I原子的键合路径。NCIplot可视化分析表明Ag+…Ag+和I*Ag的相互作用都是相互吸引的(图4CD)。通过理论计算分析配合物2和7的键临界点分布和键合路径也可以发现配合物7中I*Ag键临界点的电荷密度和拉普拉斯算子(Laplacian)远比配合物2中Ag+…Ag+键临界点的高,表明I*Ag之间的相互作用更强,这也与I+…Ag+之间的距离比Ag+…Ag+较短相符。
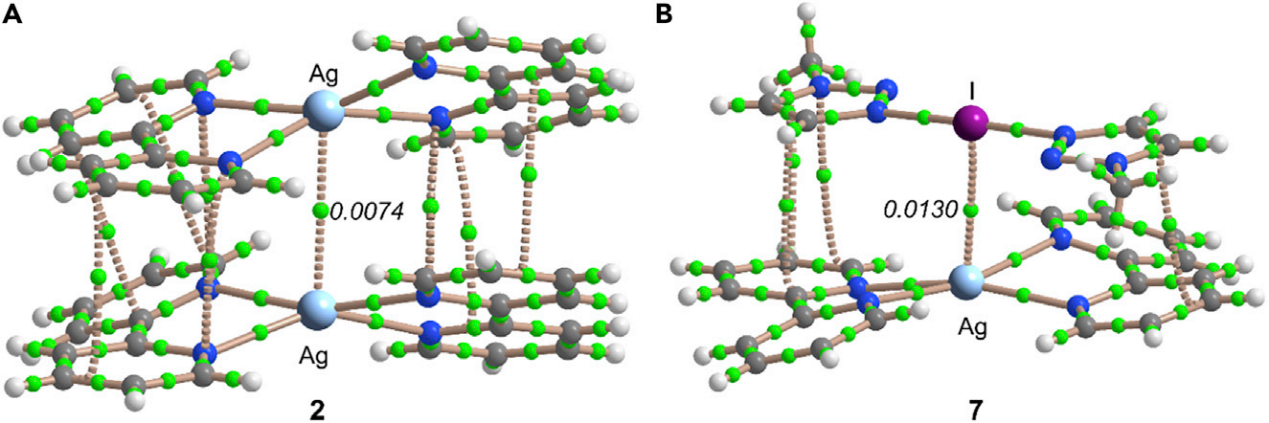
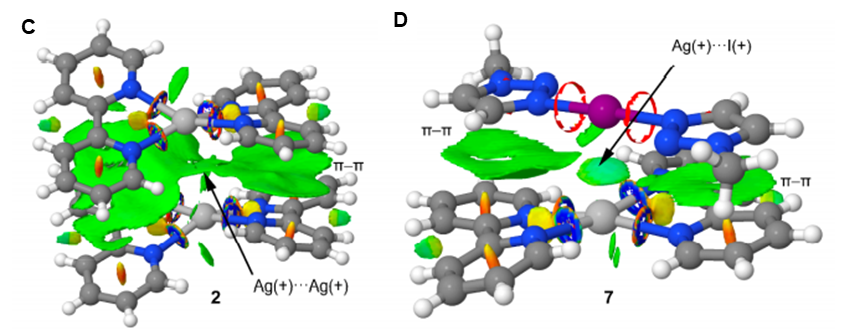
图4:A, B: 配合物2和配合物7的二聚体的键临界点(绿色球)分布和键路径(虚线对应非共价相互作用),C, D:配合物2和配合物7的NCIplot可视化分析,吸引力强度:红色<绿色<蓝色
为了研究配合物7中两种正离子结合的原因,作者基于自然键轨道理论(natural bond orbital,NBO)对配合物2和7进行了分析。结果如图5显示,化合物2中存在由Ag1的4d原子轨道向Ag2空的5s轨道提供电子。此外,也存在一个等效的“反向捐赠”4d (Ag2)→5s (Ag1)。而I+…Ag+相互作用不同于此前研究较多的Ag+…Ag+相互作用,因为仅发现了I→Ag供电子效应。配合物7的稳定能(32.10 kcal/mol)比配合物2(11.22 kcal/mol)高,这与I+…Ag+之间的距离较短有关,并与QTAIM分析结果一致。
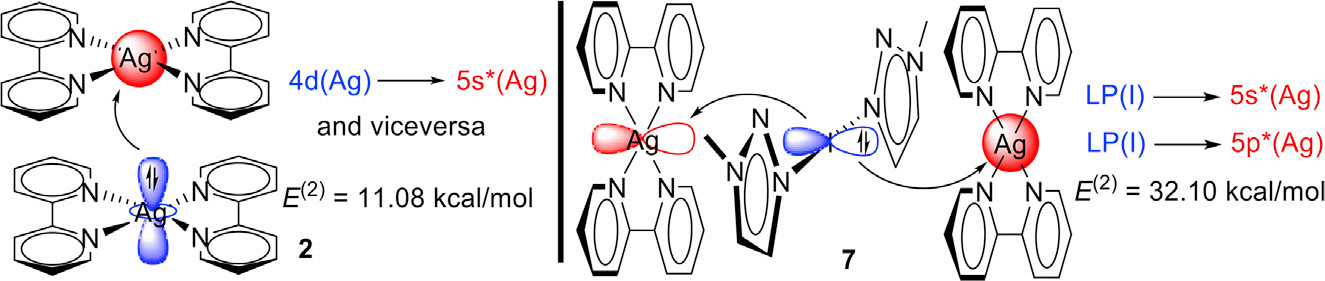
图5:用M06-2X/def2-QZVP波函数表示配合物2(左)和7(右)轨道给体-受体相互作用
结论
通过实验和理论研究,作者证明带正电荷的两个碘鎓离子以及碘鎓离子和银离子在溶液和固态下都存在较强的相互作用,I*Ag相互作用的稳定性允许通过三种不同的路线制备7,或通过简单的按1:1比例重结晶碘和银配合物,或通过中间体异感银配合[{Ag(bpy)(mtz)}2][PF6]2和碘的复分解反应得到。其中碘鎓离子作为富电子物种,而银离子作为Lewis酸,这与此前研究较多的Ag+…Ag+相互作用不同,而且前者相互作用明显更强。这些非同寻常相互作用的进一步研究,对发现新的化学反应及一些新功能化合物都有重要意义。
参考文献
1) Hassel, O., Hope, H., So¨ rensen, N.A., Dam, H., Sjo¨ berg, B., and Toft, J. (1961). Structure of the solid compound formed by addition of two molecules of iodine to one molecule of pyridine. Acta Chem. Scand. 15, 407–416.
2)Barluenga, J., Trincado, M., Rubio, E., and Gonza´ lez, J.M. (2003). IPy2BF4-promoted intramolecular addition of masked and unmasked anilines to alkynes: direct assembly
of 3-Iodoindole cores. Angew. Chem. Int. Ed. Engl. 42, 2406–2409.
3) Chalker, J.M., Thompson, A.L., and Davis, B.G. (2011). Safe and scalable preparation of Barluenga’s reagent. Org. Synth. 288–298.
4) von der Heiden, D., Vanderkooy, A., and Erde´ lyi, M. (2020). Halogen bonding in solution: NMR spectroscopic approaches. Coord. Chem. Rev. 407, 213147.
5) Lindblad, S., Mehmeti, K., Veiga, A.X., Nekoueishahraki, B., Gra¨ fenstein, J., and Erde´ lyi, M. (2018). Halogen bond asymmetry in solution. J. Am. Chem. Soc. 140, 13503–13513.
6) Karim, A., Reitti, M., Carlsson, A.-C.C., Gra¨ fenstein, J., and Erde´ lyi, M. (2014). The nature of [N–Cl–N]+ and [N–F–N]+ halogen bonds in solution. Chem. Sci. 5, 3226–3233.
7) Carlsson, A.-C.C., Uhrbom, M., Karim, A., Brath, U., Gra¨ fenstein, J., and Erde´ lyi, M. (2013). Solvent effects on halogen bond symmetry. CrystEngComm 15, 3087–3092.
8) Carlsson, A.-C.C., Gra¨ fenstein, J., Budnjo, A., Laurila, J.L., Bergquist, J., Karim, A., Kleinmaier, R., Brath, U., and Erde´ lyi, M. (2012). Symmetric halogen bonding is preferred in solution. J. Am. Chem. Soc. 134, 5706–5715.
9) Carlsson, A.-C.C., Gra¨ fenstein, J., Laurila, J.L., Bergquist, J., and Erde´ lyi, M. (2012). Symmetry of [N–X–N]+ halogen bonds in solution. Chem. Commun. 48, 1458–1460.
10) Mun˜ iz, K., Garcı´a, B., Martı´nez, C., and Piccinelli, A. (2017). Dioxoiodane compounds as versatile sources for iodine(I) chemistry. Chemistry 23, 1539–1545.
原文链接:https://doi.org/10.1016/j.chempr.2021.01.003
撰稿人:2019级博士生卫彩云
校稿人:2019级博后李晓慧
")
